
We are currently focusing on four aspects of the evolution of eukaryotic microbes:
- Assembling the tree of life with a focus on microbial lineages
- Foraminifera biodiversity and genome evolution
- Biodiversity and biogeography of testate amoebae in bogs and fens
- Genome evolution in ciliates and phylogeography of coastal choreotrich and oligotrich ciliates
Assembling the tree of life with a focus on microbial lineages
The Katzlab focuses on phylogenomics of eukaryotes. Eukaryotes, cells with nuclei, existed exclusively as microorganisms for at least 500 million years before the evolution of plants and animals. Microbial eukaryotes, also called protists, are a diverse group of organisms with a fascinating evolutionary history (e.g. multiple acquisitions of chloroplasts) and present biology (e.g. scrambled genomes). They also play an essential role in ecosystems, including in carbon fixation in marine systems. In the Katzlab, we are working to generate and analyze high throughout data to elucidate the eukaryotic tree of life with particular interest in protists. This includes investigating meiotic-specific genes in putatively-asexual lineages and in diverse microeukaryotes. Other recent papers focus on phylogeny, lateral gene transfer, and genome evolution (see below).
Foraminifera biodiversity and genome evolution
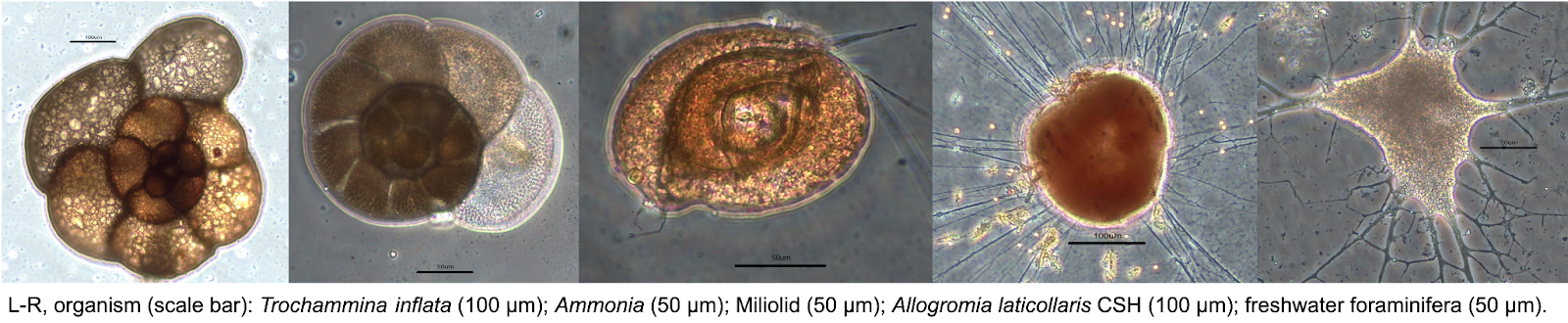
Foraminifera, or forams for short, belong to a diverse clade of marine and freshwater amoeboid protists.
Biodiversity and biogeography of testate amoebae in bogs and fens
“Everything is everywhere, but the environment selects.” – Lourens Baas Becking
Though common wisdom is that for microbes, ‘everything is everywhere,’ it is unclear how microbial lineages respond to global shifts in climate (e.g. periods of glaciation, the current warming climate). Small, single-celled organisms, like shelled amoebae in the group Arcellinida, are highly abundant in freshwater ecosystems, like low-pH bogs and fens, that are threatened by shifting trends in temperature and rainfall. Despite their size, these microorganisms can travel large distances to colonize new habitats, but the ways in which this happens are poorly understood. In the Katz lab, we use single-cell transcriptomes (to identify individuals) and lineage-specific amplicon sequencing of DNA and RNA from environmental samples (to characterize communities) to answer pressing ecological and evolutionary questions. By examining current diversity, mechanisms of dispersal, and associated habitat preferences, we can infer how changes in the environment might impact Arcellinida abundance and ability to migrate and populate new habitats in the future.
Genome evolution in ciliates and phylogeography of coastal choreotrich and oligotrich ciliates
In collaboration with George McManus, an ecologist at the University of Connecticut, we are collecting molecular data to assess the phylogeography of marine ciliates. Current ideas on protist biogeography fall into one of two camps: (1) all protists are cosmopolitan (high gene flow) and we have discovered the bulk of protist species, and (2) there are many endemic species (low gene flow) of protists yet to be discovered. Our data suggest the need to redefine this debate as we find repeated evidence of ciliate lineages with both high gene flow and high genetic diversity (e.g. Katz et al. 2005). We are now analyzing High Throughput Sequencing (HTS) data generated using primers designed to characterize SAR (Stramenopila, Alveolata, Rhizaria).
Ciliates belong to the Alveolata and are defined by the presence of their two distinct genomes, each contained within its own nucleus known as ‘germline’ micronucleus (MIC) and somatic macronucleus (MAC). The ‘germline’ micronucleus (MIC), undergoes meiosis and mitosis but is transcriptionally inactive. In contrast, the macronucleus (MAC) is the site of virtually all transcription within ciliates. The macronuclear genome develops from a zygotic genome through a series of sometimes-extensive chromosomal rearrangements, including fragmentation of chromosomes, elimination of specific sequences and amplification of the remaining chromosomes.
The Katzlab has focused on exploring patterns of chromosomal rearrangements among diverse ciliate lineages. We demonstrated that extensively fragmented genomes occur in three classes of ciliates: in addition to the well-studied class Spirotrichea, the Armophorea and Phyllopharyngea extensively fragment their MAC genome to generate ‘gene-sized’ chromosomes. For example, in these lineages the ~1.6kb a-tubulin gene resides on a chromosome 1.8-2.2 kb in length. The appearance of several lineages containing extensively fragmented genomes suggests multiple origins of gene-sized MAC chromosomes.
Having established that extensively processed genomes evolved multiple times in ciliates, we are now characterizing the mechanisms of genome rearrangements from diverse ciliates. Most recently, we have demonstrated that alternative processing of germline regions underlies gene family evolution in ciliates. Our analyses suggest that the dimorphic nature of ciliate genomes enables ciliates to cross the ‘valleys’ in the adaptive landscape of protein evolution— ciliates may be able to maintain deleterious copies of paralogs in their MICs while ‘hiding’ them from selection in their somatic macronucleus. Processed MAC genomes are as following: (1) break up linkage groups (fate of paralogs less affected by fate of other polymorphisms on linked chromosomes); (2) allow assortment of paralogs in the MAC but not in the MIC (deleterious mutations may be hidden in the MAC even though they are present in the MIC); and (3) redefine ‘genetic load’ through differential amplification of MAC chromosomes (may make it relatively inexpensive for ciliates to carry duplicated genes).